- Home
- I am a …
- Resources
- Collections
- Post-lockdown teaching support
- Remote teaching support
- Starters for ten
- Screen experiments
- Assessment for learning
- Microscale chemistry
- Faces of chemistry
- Classic chemistry experiments
- Nuffield practical collection
- Anecdotes for chemistry teachers
- More …
- Literacy in science teaching
- Climate change and sustainability
- Alchemy
- On this day in chemistry
- Global experiments
- PhET interactive simulations
- Chemistry vignettes
- Context and problem based learning
- Journal of the month
- Chemistry and art
- Classic chemistry demonstrations
- In search of solutions
- In search of more solutions
- Creative problem-solving in chemistry
- Solar spark
- Chemistry for non-specialists
- Health and safety in higher education
- Analytical chemistry introductions
- Exhibition chemistry
- Introductory maths for higher education
- Commercial skills for chemists
- Kitchen chemistry
- Journals how to guides
- Chemistry in health
- Chemistry in sport
- Chemistry in your cupboard
- Chocolate chemistry
- Adnoddau addysgu cemeg Cymraeg
- The chemistry of fireworks
- Festive chemistry
- Collections


- Education in Chemistry
- Teach Chemistry
- Events
- Teacher PD
- Enrichment
- Our work
- More from navigation items
Close menu
- Home
- I am a …
-
Resources
- Back to parent navigation item
- Resources
- Primary
- Secondary
- Higher education
- Curriculum support
- Practical
- Analysis
- Literacy in science teaching
- Periodic table
- Climate change and sustainability
- Careers
- Resources shop
-
Collections
- Back to parent navigation item
- Collections
- Post-lockdown teaching support
- Remote teaching support
- Starters for ten
- Screen experiments
- Assessment for learning
- Microscale chemistry
- Faces of chemistry
- Classic chemistry experiments
- Nuffield practical collection
- Anecdotes for chemistry teachers
- More …
- Literacy in science teaching
- Climate change and sustainability
- Alchemy
- On this day in chemistry
- Global experiments
- PhET interactive simulations
- Chemistry vignettes
- Context and problem based learning
- Journal of the month
-
Chemistry and art
- Back to parent navigation item
- Chemistry and art
- Techniques
- Art analysis
- Pigments and colours
- Ancient art: today's technology
- Psychology and art theory
- Art and archaeology
- Artists as chemists
- The physics of restoration and conservation
- Cave art
- Ancient Egyptian art
- Ancient Greek art
- Ancient Roman art
- Classic chemistry demonstrations
- In search of solutions
- In search of more solutions
- Creative problem-solving in chemistry
- Solar spark
- Chemistry for non-specialists
- Health and safety in higher education
- Analytical chemistry introductions
- Exhibition chemistry
- Introductory maths for higher education
- Commercial skills for chemists
- Kitchen chemistry
- Journals how to guides
- Chemistry in health
- Chemistry in sport
- Chemistry in your cupboard
- Chocolate chemistry
- Adnoddau addysgu cemeg Cymraeg
- The chemistry of fireworks
- Festive chemistry
- Education in Chemistry
- Teach Chemistry
- Events
- Teacher PD
- Enrichment
- Our work








Analysis resources
Discover resources to support you as you teach the principles and practice of spectroscopy and other analytical methods

A model of mass spectrometry
Use this modelling activity to explore your students’ understanding of the process of fragmentation in mass spectrometry.
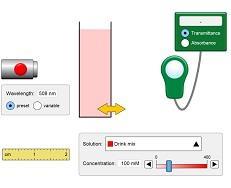
Beer's Law simulation
In association with PhET Interactive Simulations University of Colorado Boulder
Bring this theoretical principle to life.
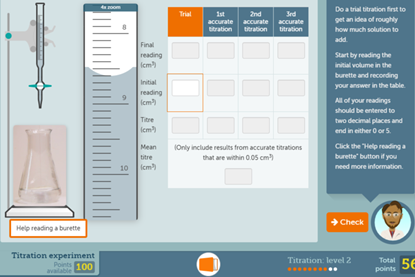
Titration screen experiment
Give students the opportunity to conduct their own titration experiment on a computer or tablet. This resource also includes a redox titration experiment.

Analysis
Develop your students’ knowledge and understanding of analytical techniques with the help of games, activities, practicals and demonstrations.

Infrared Spectrometer
Watch an illuminating six-minute video explaining how infrared spectroscopy works.

Chromatography challenge | 16–18 years
Explore analytical techniques and their applications with a chromatography investigation and research activity
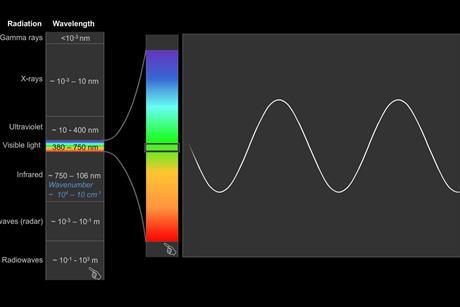
Introduction to spectroscopy
Get back to basics with this primer on the principles of spectroscopic techniques, including infrared (IR), ultraviolet-visible (UV-vis) and nuclear magnetic resonance (NMR). To make it even easier, each technique has clear explanations and descriptions supported by animations.